ICCR datasets are developed under a quality framework which dictates both how the datasets look as well as what should be included. Each dataset has been developed by an international panel, the Dataset Authoring Committee (DAC), and includes Core and Non-core elements. The roles and responsibilities of the DAC and others in the dataset development process are outlined in Roles and Responsibilities for the ICCR dataset development process.
Core elements are those which are essential for the clinical management, staging or prognosis of the cancer. These elements will either have evidentiary support at Level P2 or above, based on prognostic factors in the Hierarchy of Research Evidence for Tumour Pathology table below:
World Health Organization Hierarchy of Research Evidence for Tumour Pathology.
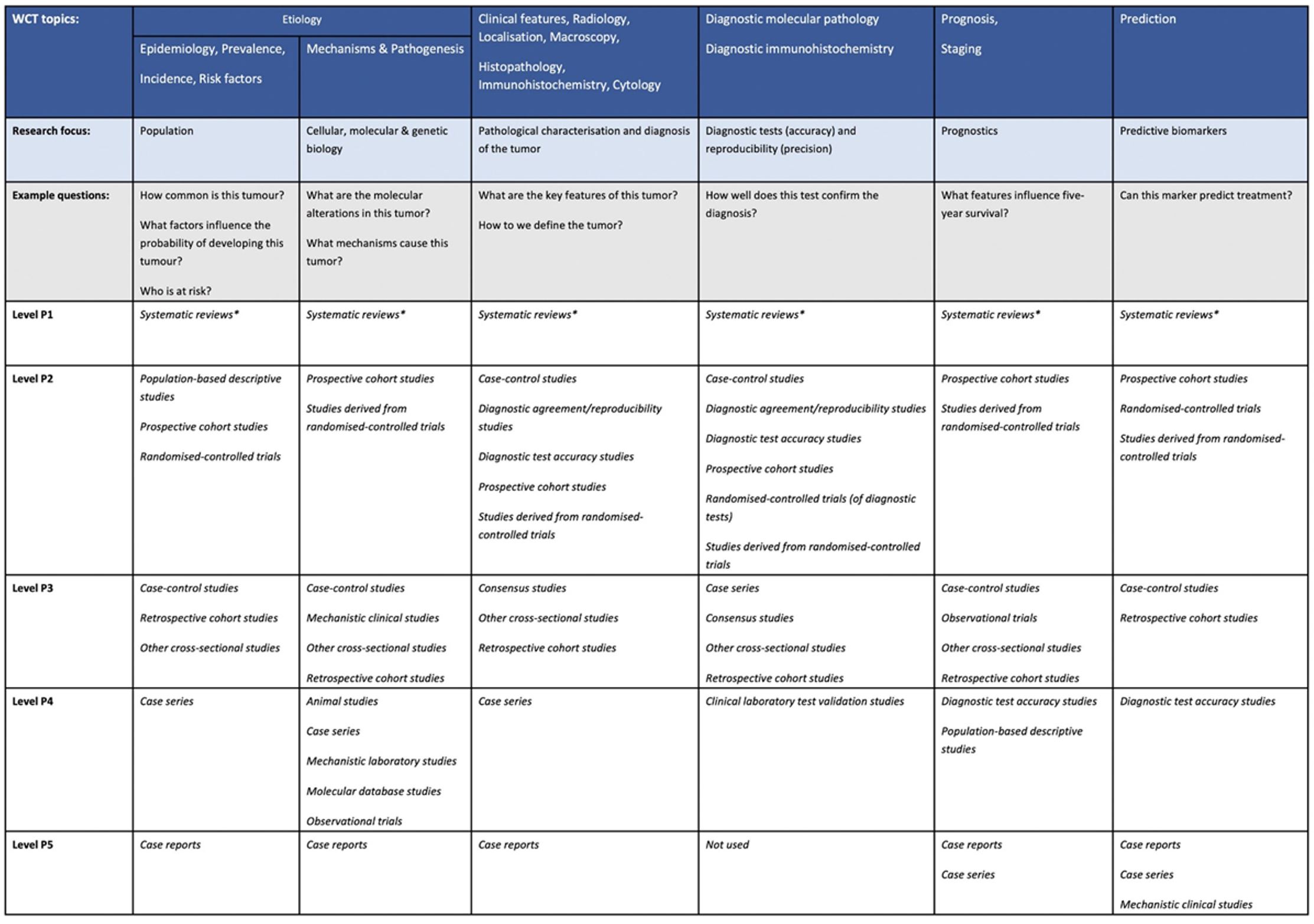
The hierarchy is arranged at the top level to map to the various sections of the World Health Organization Classification of Tumours, that is, matching chapter headings/subheadings as far as possible to cover all topics discussed in the series. The various headings are grouped into similar research topics of interest, such as tumour characteristics, prognostics, and so forth. The levels presented below are in order of robustness, that is, level P1 having the greatest confidence and level P5 the lowest. Each P level for each subsection contains the list of evidence types that rank at that level (in alphabetical order, no intralevel ranking implied). Most types of evidence can be thought of as ‘studies’ but not all. Studies of rare tumours (defined here as those with an incidence of <1/million population/annum) are automatically upgraded to 1 level. This would include case reports that may be the only unique diagnosis of its type in the literature. Large sample size level P2 studies should be upgraded to level P1. This table should be used in conjunction with the glossary (Supplementary File 2) as the terms used may be different from those that some readers may be used to. * Systematic reviews predominantly including level P2 studies. Systematic reviews of predominantly level P3-P5 studies are placed in level P2. Rapid reviews are placed in level P2.
From: Colling R, Indave I, Del Aguila J, Jimenez RC, Campbell F, Chechlińska M, Kowalewska M, Holdenrieder S, Trulson I, Worf K, Pollán M, Plans-Beriso E, Pérez-Gómez B, Craciun O, García-Ovejero E, Michałek IM, Maslova K, Rymkiewicz G, Didkowska J, Tan PH, Md Nasir ND, Myles N, Goldman-Lévy G, Lokuhetty D and Cree IA (2024). A New Hierarchy of Research Evidence for Tumor Pathology: A Delphi Study to Define Levels of Evidence in Tumor Pathology. Mod Pathol 37(1):100357.
In rare circumstances, where level P2 evidence is not available an element may be made a Core element where there is unanimous agreement in the DAC. An appropriate staging system e.g., Pathological TNM staging would normally be included as a Core element.
Non-core elements are those which are unanimously agreed should be included in the dataset but are not supported by level P2 evidence. These elements may be clinically important and recommended as good practice but are not yet validated or regularly used in patient management.
The dataset development process is outlined in the Guidelines for the Development of ICCR Datasets document and in the figure below:
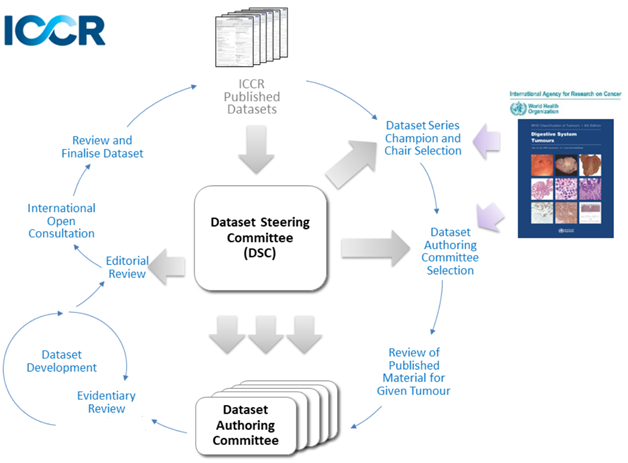
To ensure a consistent approach to the content of ICCR datasets, terms and their recommended usage have been defined and are available in the ICCR Harmonisation Guidelines.
These documents will be updated periodically in order to maintain currency and to take advantage of improvements in process and agreement in terminology that will be achieved as the collaborative process progresses.
There is a close interdependency between cancer reporting datasets and the World Health Organization (WHO) Classification of Tumours ‘Blue Books’, and therefore the ICCR has made a commitment to publish datasets in synchrony with the WHO updates. Therefore, new datasets will generally be developed in synchrony with the WHO updates, however, changes to other dependent publications such as TNM staging or International Federation of Gynaecology and Obstetrics (FIGO) staging will also trigger development of new datasets. Additional datasets may be scheduled for development if a specific need arises and resources are available.
ICCR cancer datasets will be scheduled for update in synchrony with revisions to the WHO Classification of Tumours. Updates before the date of formal review may also be undertaken as a result of errors, changes to staging systems, or significant changes in clinical or diagnostic evidence or management related to a specific cancer for example.
The process of updating ICCR cancer datasets is described in the Guidelines for the Development of ICCR Datasets document.